Diagnosing unresolved Stargardt disease using retina-specific splicing isoforms obtained from retinal organoids
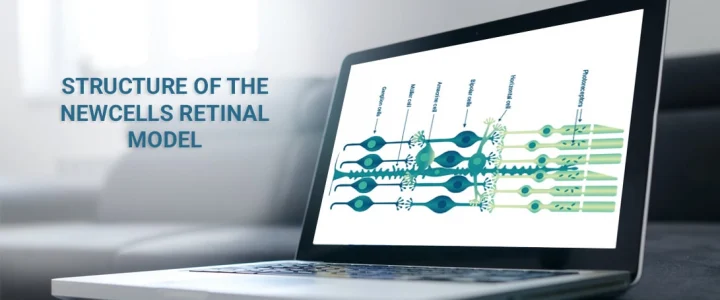
At Newcells Biotech, we specialise in the large-scale production of retinal organoids for the purpose of in vitro toxicology efficacy studies and disease modelling. Our differentiation protocols generate organoids which are fully stratified with the presence of all major retinal cells including rod and cone photoreceptors, retinal ganglion cells (RGCs), bipolar cells, horizontal cells, amacrine cells and Müller glial cells.
Throughout the last decade, a number of 2D models have been superseded by the development of 3D mini-organs, also known as organoids. These organoids have been shown to closely recapitulate in vivo biology in an in vitro context, with significantly higher fidelity than previous 2D systems.
The first report of functionality within retinal organoids came from the Canto-Soler group, who achieved viable long-term culture and the development of photoreceptors with rudimentary outer segments; this work was followed by similar work by others.1,2,3 Outer segments are a critical component of phototransduction. Indeed, the early structures found in these long-term organoid cultures displayed evidence of photosensitivity via the expression of key phototransduction-related molecules.
These studies showed proof-of-concept for in vitro modelling of an in vivo complex retinal system; however, the resulting organoid yields were often quite variable. This problem was addressed through our NC3Rs–sponsored collaboration with Professor Lako at Newcastle University which resulted in the development of a protocol for the efficient production of retinal organoids in a 96 well-plate format.1 By adapting this protocol to multi-well plate format, the production of retinal organoids could be significantly upscaled, with the added benefit of less physical maintenance. This provides an excellent platform for in vitro drug testing.
One of our key areas of interest at Newcells Biotech is to utilise these organoids for diagnostic research purposes. We are a partner in the EU StarT consortium, which focuses on genetic diagnosis, pathological mechanisms, and treatment of the inherited blinding disorder Stargardt Disease.
Stargardt disease is the leading cause of inherited maculopathy and is caused by mutations in the ABCA4 gene.4 It has an overall incidence of 1:8000-10,000 individuals.5 Symptoms of this disease begin to manifest within the first two decades of life and progress over time to a stage where individuals become completely centrally blind. This means that affected persons can no longer read or write, recognize family & friends, drive or watch television. Therefore, this disease severely impacts quality of life. Furthermore, to date, there is no successful treatment for this disease, highlighting the need for increased research into the mutational spectrum and associated pathology.6
As a recessive condition, in up to 72% of sequenced cases, biallelic variants are identified in ABCA4.7 However, this leaves around 30% of STGD1 genetically unsolved, with either one or no ABCA4 variants despite phenotypically displaying symptoms of the disease. It is extremely important to have a genetic diagnosis of an inherited disorder so that an affected individual can access appropriate counselling and also enlist in any clinical trials for potentially sight-saving therapies.
Monoallelic cases have been of great interest in STGD1 research. This is owing to the identification of deep-intronic ABCA4 variants in a number of cases that have been shown to alter the conventional splicing patterns in the retina.8-11 RNA defects appear to play a more prominent role in disease pathology than previously expected. However, obtaining relevant ABCA4 RNA transcripts from affected individuals is not an easy task. ABCA4 is expressed uniquely in the photoreceptor cells of the retina, where site-specific splicing programs also occur.12 Therefore, to accurately identify pathological RNA defects, RNA would have to be isolated from an affected individual’s retina, which is clearly not possible in living patients.
To bypass this issue, we generate iPSCs from monoallelic STGD1 patients using an RNA-based transfection method,13 which has been validated in our labs. We then culture retinal organoids for up to 220 days, so that they have mature photoreceptors from which we can isolate ABCA4 RNA and carry out gene expression and sequencing analyses. It has previously been shown that RNA isolated from iPSC-derived photoreceptor precursor cells could faithfully detect pathological RNA defects that were previously undetected using splicing algorithms.14
For genomic sequencing, we are using long-read sequencing technology to compare monoallelic ABCA4 transcripts against unaffected individuals to negate the use of reference genomes which are often generated using short-read sequencing and splicing inference tools. Long-read sequencing, although more costly, is extremely accurate at calling nucleotide bases, as well as providing long subreads which span across intron/exon junctions negating the dependence on contig assembly after sequencing.15
Through this experimental approach, we hope to identify the missing inheritance in these monoallelic cases, thereby providing a genetic diagnosis as well as expanding current insights into the ABCA4 RNA landscape.
References
1.Hallam, D. et al. Human-Induced Pluripotent Stem Cells Generate Light Responsive Retinal Organoids with Variable and Nutrient-Dependent Efficiency. STEM CELLS 36, 1535-1551 (2018).
2.Cowan, C.S. et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 182, 1623-1640.e34 (2020).
3.Zhong, X. et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun 5, 4047 (2014).
4.Allikmets, R. et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 15, 236-46 (1997).
5.Tsang, S.H. Sharma, T. Stargardt Disease. Adv Exp Med Biol 1085, 139-151 (2018).
6.Tanna, P., Strauss, R.W., Fujinami, K. & Michaelides, M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol 101, 25-30 (2017).
7.Zernant, J. et al. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb Mol Case Stud 4(2018).
8.Bax, N.M. et al. Heterozygous deep-intronic variants and deletions in ABCA4 in persons with retinal dystrophies and one exonic ABCA4 variant. Hum Mutat 36, 43-7 (2015).
9.Runhart, E.H. et al. Late-Onset Stargardt Disease Due to Mild, Deep-Intronic ABCA4 Alleles. Investigative Ophthalmology & Visual Science 60, 4249-4256 (2019).
10.Sangermano, R. et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T→C Mutation in Stargardt Disease. Ophthalmology 123, 1375-85 (2016).
11.Bauwens, M. et al. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum Mutat 36, 39-42 (2015).
12.Murphy, D., Cieply, B., Carstens, R., Ramamurthy, V. & Stoilov, P. The Musashi 1 Controls the Splicing of Photoreceptor-Specific Exons in the Vertebrate Retina. PLoS Genet 12, e1006256 (2016).
13.Chichagova, V., Sanchez-Vera, I., Armstrong, L., Steel, D. & Lako, M. Generation of Human Induced Pluripotent Stem Cells Using RNA-Based Sendai Virus System and Pluripotency Validation of the Resulting Cell Population. Methods in molecular biology (Clifton, N.J.) 1353, 285-307 (2016).
14.Albert, S. et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. American journal of human genetics 102, 517-527 (2018).
15.Gonzalez-Garay, M.L. Introduction to Isoform Sequencing Using Pacific Biosciences Technology (Iso-Seq). in Transcriptomics and Gene Regulation (ed. Wu, J.) 141-160 (Springer Netherlands, Dordrecht, 2016).
Share on social media:
Don't miss out on our latest innovations: follow us on Linkedin
Newcells Biotech
27th October, 2020
Retina
